
Welcome to Nanjing YDB Medical Technology Co., Ltd
Products
LeSifterTM
![]()
Left Atrial Appendage Closure Device
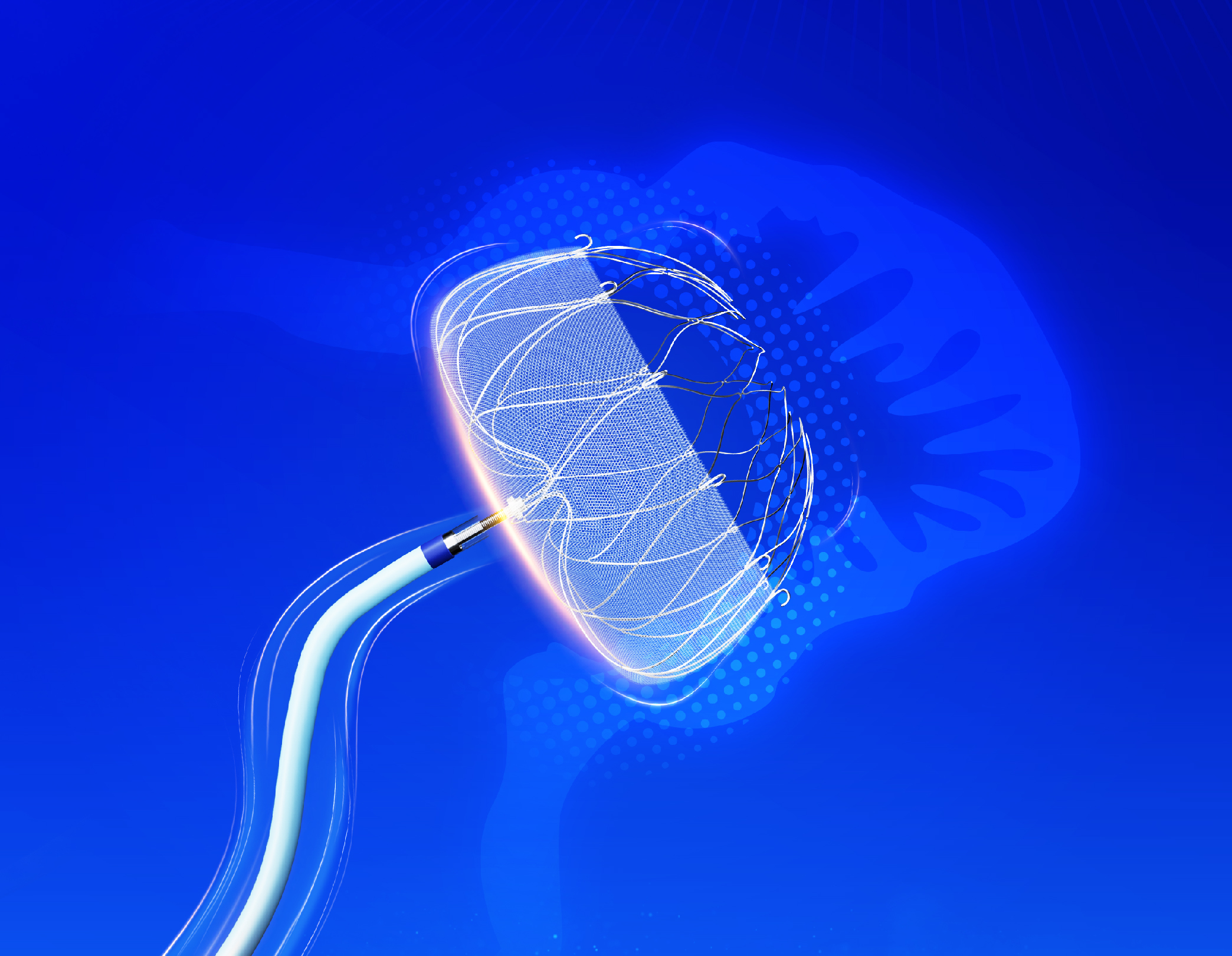
Device Description
The left atrial appendage (LAA) Closure Device consists of the LAA occluder and a Access System (Delivery System and Guiding System), where the occluder is pre-loaded into the Delivery System. The Guiding System include a Dilation Tube and a Guiding sheath. The Occluder and Delivery system can be introduced through the femoral vein and passed across the atrial septum to reach the left atrium for placement of the occluder into the left atrial appendage. The occluder is made of self-expanding nitinol alloy material, with its outer surface covered by a porous mesh. The product is available in 5 size options, ranging from 20mm to 35mm. The appropriate size of the occluder is selected by measuring the size of the left atrial appendage orifice using fluoroscopy (fluoroscopic imaging) and transesophageal echocardiography (TEE).
The LAA occluder is designed for permanent implantation at the orifice (opening) of the left atrial appendage or slightly distal to it, in order to intercept thrombus generated within the left atrial appendage. The implantation procedure is performed in the catheterization lab under local or general anesthesia.
Intended Use
LeSifterTM is intended for percutaneous, transcatheter closure of the left atrial appendage.
Indications For Use
The LesifterTM Device is indicated to reduce the risk of thromboembolism from the left atrial appendage in patients with
non-valvular atrial fibrillation who:
• Are at increased risk for stroke and systemic embolism based on CHA2DS2-VASc1 scores and are recommended for
anticoagulation therapy;
• Are deemed by their physicians to be suitable for anticoagulation therapy; and
• Have an appropriate rationale to seek a non-pharmacologic alternative to anticoagulation therapy, taking into account the safety and effectiveness of the device compared to anticoagulation therapy.
Contraindications
Do not use the LeSifterTM Device if:
• Intracardiac thrombus is present.
• An atrial septal defect repair or Occluder is present.
• A patent foramen ovale repair or Occluder is present.
• The LAA anatomy will not accommodate a Occluder.
• The patient has a known hypersensitivity to any portion of the device material or the individual components such that the use of the LeSifterTM Device is contraindicated.
• Any of the customary contraindications for other percutaneous catheterization procedure (e.g., patient size too small to accommodate TEE probe or required catheters) or conditions (e.g., active infection, bleeding disorder) are present.
• There are contraindications to the use of anticoagulation therapy, aspirin, or P2Y12 inhibitor.
Intended User:
Intended users of the LesifterTM left atrial appendage Occluder are interventional cardiologists and/or electrophysiologists who are proficient in percutaneous procedures, transseptal procedures, the imaging modality utilized.
Warining
• Contents supplied STERILE using an ethylene oxide (EO) process. Do not use if sterile barrier is damaged.
If damage is found, call your YDB Medical representative.
• Implantation of the Device should only be performed by interventional cardiologists and/or electrophysiologists who are proficient in percutaneous procedures, transseptal procedures, the imaging modality utilized
• For single use only. Do not reuse, reprocess, or resterilize.
• This device has not been studied in pregnant or breastfeeding women. Careful consideration should be given to use of the Occluder in pregnant and/ or breastfeeding women due to the risk of significant exposure to x-rays and the use of anticoagulation medication.
• Device selection should be based on accurate LAA measurements obtained using transesophageal or intracardiac echocardiographic imaging guidance in multiple views to avoid improper Occluder sizing. For TEE recommended in multiple angles [e.g., 0°, 45°, 90°, 135°]; For ICE imaging, visualization of the LAA is recommended with the following anatomical structures: aortic valve (short-axis), mitral valve (long-axis), and pulmonary artery (short-axis), to assess the minimum and maximum diameter of the LAA ostium.
• Do not release (i.e., unscrew) the occluder from the core wire unless all release criteria are satisfied to avoid suboptimal results.
• Potential for Occluder embolization exists with cardioversion < 30 days following Occluder implantation; verify Occluder position after cardioversion during this period.
Adverse Events
Potential adverse events which may be associated with the use of a left atrial appendage Occluder or implantation procedure include but are not limited to:
• Air embolism
• Airway trauma
• Allergic reaction to the contrast media, anesthetic, WATCHMAN Implant material, or medication• Altered mental status
• Anemia requiring transfusion
• Anesthesia risks
• Angina
• Anoxic encephalopathy
• Arrhythmias
• Atrial septal defect
• Bruising, hematoma, or seroma near the catheter insertion site
• Cardiac perforation
• Chest pain/discomfort
• Confusion post-procedure
• Congestive heart failure
• Contrast-related nephropathy
• Cranial bleed
• Death
• Decreased hemoglobin
• Deep vein thrombosis
• Device embolism
• Device fracture
• Device thrombosis
• Edema
• Embolism
• Excessive bleeding
• Fever
• Fistula
• Groin pain
• Groin puncture bleed
• Hematuria
• Hemoptysis
• Hypotension
• Hypoxia
• Improper wound healing
• Inability to reposition, recapture, or retrieve the device
• Infection/pneumonia
• Interatrial septum thrombus
• Intratracheal bleeding
• Major bleeding requiring transfusion
• Misplacement of the device/improper seal of the appendage/movement of device from appendage
• wall
• Myocardial erosion
• Myocardial infarction
• Nausea
• Oral bleeding
• Pericardial effusion/tamponade
• Pleural effusion
• Prolonged bleeding from a laceration
• Pseudoaneurysm
• Pulmonary edema
• Radiation injury
• Renal failure
• Respiratory insufficiency/failure
• Stroke - Hemorrhagic
• Stroke - Ischemic
• Surgical removal of the device
• TEE complications (e.g., throat pain, bleeding, esophageal trauma)
• Thrombocytopenia
• Thrombosis
• Transient ischemic attack (TIA)
• Valvular or vascular damage
• Vasovagal reactions
There may be other potential adverse events that are unforeseen at this time.
Patient selection for treatment
the LeSifterTM Device is an option that may be considered in patients to reduce the risk of cardioembolism from the LAA.Selection among available treatment options must first take into account whether anticoagulation is indicated to reduce the risk of stroke based on CHA2DS2-VASc scores. Next, in a patient who is deemed by their physicians to be suitable for anticoagulation therapy, physicians and patients should consider the rationale for implantation of the LeSifterTM Device as an alternative to long-term anticoagulation therapy. Specific factors may include one or more of the following:
• A history of major bleeding while taking anticoagulation therapy.
• The patient’s prior experience with oral anticoagulation (if applicable).
• A medical condition, occupation, or lifestyle placing the patient at high risk of major bleeding secondary to trauma. Some studies of patients with a history of falls, or at risk for falls and head trauma, have shown that the benefits of anticoagulation therapy to reduce the risk of stroke outweigh the risk of major, life-threatening bleeding. An individualized benefit and risk assessment should be made in such patients.
• The presence of indication(s) for long-term anticoagulation therapy, other than non-valvular atrial fibrillation (e.g. mechanical heart valve, hypercoagulable states, recurrent deep venous thrombosis).
Reuse Warning
For single use only. Do not reuse, reprocess, or resterilize. Reuse, reprocessing, or resterilization may compromise the structural integrity of the device and/or lead to device failure which, in turn, may result in patient injury, illness, or death. Reuse, reprocessing, or resterilization may also create a risk of contamination of the device and/or cause patient infection or cross-infection, including, but not limited to, the transmission of infectious disease(s) from one patient to another. Contamination of the device may lead to injury, illness, or death of the patient.
MRI Safety information
Non-clinical testing has demonstrated that the LeSifterTM device is MR Conditional.
A patient with the LeSifterTM device can be safely scanned in an MR system under the following conditions:
- Static magnetic fields of 3.0 Tesla (3.0T)
- Maximum spatial gradient field of 19 T/m (1900 G/cm)
- Maximum MR system reported, whole-body averaged specific absorption rate (SAR) of 2.0 W/kg (normal operating mode)
Under the scan conditions defined above, the device is expected to produce a maximum temperature rise of less than or equal to 1.6°C after 15 minutes of continuous scanning.
In non-clinical testing, the image artifact caused by the device extends radially up to 10 mm from the device when imaged with a gradient echo pulse sequence in a 3.0T MR system
Operation Instractions
Preparation
A baseline measurement by means of appropriate imaging modality (either cardiac CT or TEE) should be performed to verify if a LeSifterTM Device can be implanted and preselect the appropriate occluder size.
1.Perform the following through multiple imaging views:
• Measure the LAA length and width at the ostium.
• Assess LAA size/shape,number of lobes in LAA,and location of lobes to ostiun.
• Confirm the absence of thrombus.
Note:If using TEE,measure the LAA ostium at approximately these angles as anatomy permits:
• At 0°measure from coronary artery marker to a point approximately 2 cm from tip of the ‘’limbus.’’
• At 45°measure from the top of the mitral valve annulus to a point approximately 2 cm from tip of the ‘’limbus.’’
• At 90°measure from the top of the mitral valve annulus to a point approximately 2 cm from tip of the ‘’limbus.’’
• At 135°measure from the top of the mitral valve annulus to a point approximately 2 cm from tip of the ‘’limbus.’’
2.Determine the greatest width (i.e.diameter) measurement.
3.Record LAA ostium width and LAA depth measurements. Use Table 1 as a guide for size selection.
Measured maximum LAA ostium width must be ≥14.0mm and≤32.0mm to accommodate available Occluder sizes.
Note:Successful device sizing is dependent on multiple imaging views.
Note:The maximum LAA ostium width and depth measurements determine Occluder size selection.
Table 1 :LeSifterTM Device Selection
|
Model |
Max LAA Ostium Width(mm) |
Occluder Size(mm) |
|
YFDQ-20-1/ YFDQ-20-2 |
14~18 |
20 |
|
YFDQ-24-1/ YFDQ-24-2 |
17~21 |
24 |
|
YFDQ-27-1/ YFDQ-27-2 |
19~24 |
27 |
|
YFDQ-31-1/ YFDQ-31-2 |
22~28 |
31 |
|
YFDQ-35-1/ YFDQ-35-2 |
25~32 |
35 |
Procedure
Equipment Needed for implantation Procedure
• Venous Introducer (optional)
• Standard transseptal access system
• 0.035 in (0.89mm) guidewire (exchange length support)
• 5F(1.7mm) or 6F(2.0mm) angioraphic pigtail catheter
Implantation Procedure
Note: Patient should start aspirin prior to scheduled procedure and continue daily.
Note: Fluoroscopic (fluoro) and echocardiographic imaging should be used when implanting the device.
Note: Patients should be fully heparinizad throughout the procedure with a recommended minimum active clotting time (ACT) of 200 seconds-300 seconds after transseptal puncture.
1.Use standard percutaneous techniques to puncture femoral vein and insert 0.035 in (0.89 mm) guidewire and vessel Dilation Tube. Use a standard, commercially available transseptal access system to cross inter-atrial septum.
2.Exchange crossing sheath with exchange length extra support 0.035 in (0.89 mm) guidewire. Position guidewire in left upper pulmonary vein (LUPV) or loop in left atrium.
3.Prepare a Guiding System.
Note: Inspect sterile package and Guiding System prior to use. If sterile barrier, labeling, packaging, or device have been compromised in any way, DO NOT USE.
A. Remove Guiding Sheath and Dilation Tube from package under sterile conditions.
B. Inspect prior to use to ensure no damage.
C. Flush Guiding Sheath and Dilation Tube with saline prior to use.
D. Ensure hemostasis valve is fully open. Insert Dilation Tube into hemostasis valve of Guiding Sheath until the two snap together.
4.Advance a Guiding System over guidewire into left atrium (LA). As the Guiding Sheath nears center of LA, unsnap the Guiding Sheath from the Dilation Tube, hold the Dilation Tube, and advance the Guiding Sheath into initial position in LA or ostium of LUPV.
Precaution: Use caution when introducing Guiding System to prevent damage to cardiac structures.
5.Ensure hemostasis valve is fully open, remove Dilation Tube and guidewire, leaving Guiding Sheath in LA or LUPV. Allow back bleed to minimize potential for introducing air before tightening valve. Flush the Guiding Sheath with saline.
If continued back bleed is observed from the valve after the Dilation Tube is removed despite attempting to close it, loosen the valve cap (counter clockwise rotation) until the cap spins freely. Then re-attempt closure of the valve while exerting gentle forward pressure on the valve cap during closure (clockwise rotation) to ensure proper engagement of the valve thread. While these steps are being undertaken, manual occlusion of the valve opening using a gloved finger is recommended to minimize blood loss.
Note: These steps may be repeated, if necessary. However, if this does not mitigate the blood leak, the user should remove and replace the Guiding Sheath before proceeding with the procedure.
6. Carefully advance pigtail catheter through Guiding Sheath into distal portion of the LAA under fluoro guidance. Obtain angiographic views of the LAA.
Note: If user notices kink in Guiding Sheath , user should remove and replace Guiding Sheath before proceeding with procedure.
Note: Record multiple angles on cine with contrast prior to advancing the Guiding Sheath into LAA. Use fluoro guidance while advancing pigtail catheter and while advancing the Guiding Sheath . Stop if resistance is felt.
7. Confirm LAA size and confirm appropriate occluder model. There is clinical evidence to support the use of TEE or intracardiac echocardiography (ICE) and fluoroscopy to guide LAAC implantation.
A. Perform the following through multiple imaging views:
• Measure the LAA length and width at the ostium.
• Assess LAA size/shape, number of lobes, and location of lobes relative to the ostium.
• Confirm the absence of thrombus.
Note: If using TEE, measure the LAA ostium at approximately these angles as anatomy permits:
• at 0° measure from coronary artery marker to a point approximately 2 cm from tip of the “limbus.”
• at 45° measure from top of the mitral valve annulus to a point approximately 2 cm from tip of the “limbus.”
• at 90° measure from top of the mitral valve annulus to a point approximately 2 cm from tip of the “limbus.”
• at 135° measure from top of the mitral valve annulus to a point approximately 2 cm from tip of the “limbus.”
Note: In the ICE-LAA study, physicians obtained at least two orthogonal (short axis and long axis) views of the LAA when using ICE from the left atrium to document the position, compression, leak, and record the tug test to comply with the PASSTM (Position, Anchor, Size, and Seal) device release criteria (refer to Step 16). These two views were further defined as:
• Short-Axis View (also called mid LA/PV view): with the ICE probe positioned in the mid LA, near or at the ostium of the left superior pulmonary vein. This resembles the 0-90° view on TEE.
• Long-Axis View (also called supra mitral view): a posterior flex, 90° rotation, and slight advancement of the probe toward the superior aspect of the mitral valve will delineate the long axis view of the LAA, which resembles the 90-135° view on TEE.
If using ICE imaging, visualize the LAA with the following anatomical structures: aortic valve (short-axis), mitral valve (long-axis), and pulmonary artery (short-axis), to assess the LAA anatomy, as well as the PASS criteria.
B. Confirm occluder based on maximum LAA ostium width recorded. Use Table 1 as a guide.
Note: LAA anatomy should accommodate a single occluder as described in Table 1.
Note: These values are based on TEE and can be utilized with ICE. Other imaging modalities may vary.
8. Prepare Delivery System.
A. Check the temperature exposure indicator on the pouch label to confirm that the product has not been compromised. See Warnings section.
B. Remove Delivery System under sterile conditions.
Note: Inspect sterile package and Delivery System prior to use. If sterile barrier, labeling, packaging, or device have been compromised in any way, DO NOT USE.
Note: Delivery sheath hemostasis valve is packaged closed.
Precaution: Use caution when manipulating the Delivery System. Excessive counterclockwise rotation of the deployment knob or Delivery System hub independent from the rest of the Delivery System can cause premature implant detachment.
C. Inspect prior to use to ensure there is no damage to hemostasis valve, catheter connections, or Occluder (through Delivery System). Confirm the Occluder is positioned completely inside the Delivery System.
Note: If Occluder extends outside the Delivery System, DO NOT USE.
D. Loosen the hemostasis valve and move the deployment knob away from the hemostasis valve to ensure the Occluder and the core wire assembly move freely. While holding the Delivery System straight, position the distal tip of the Occluder so that it is aligned with the Delivery System distal marker band.
E. Flush the Delivery System and hemostasis valve with saline, to ensure removal of all air. Then close the hemostasis valve to maintain fluid throughout system during handling.
Note: If aspirating the Delivery System during flushing, do so slowly and with limited force, to prevent the formation of air bubbles.
9. With the pigtail in the LAA, the Guiding Sheath tip position and orientation may be carefully adjusted in the LAA as required to engage the target LAA lobe or location for deployment. Slowly remove pigtail catheter.
10. Loosen hemostasis valve of Guiding Sheath , allowing back bleed before inserting the prepped Delivery System. Apply positive pressure saline to the Delivery System flush port during introduction into Guiding Sheath to obtain a wet-to-wet connection.
Note: Tightening theGuiding Sheath hemostasis valve onto the WATCHMAN FLX Pro Delivery System may damage the Delivery System.
11. To avoid introduction of air, slowly advance Delivery System into Guiding Sheath under fluoro guidance.
Precaution: Use caution when introducing Delivery System to prevent damage to cardiac structures.
12. Under fluoro guidance, align Delivery System distal marker band with most distal marker band on
Guiding Sheath. Once marker bands are aligned, stabilize Delivery System, retract Guiding Sheath , and
snap together to create the Guiding Sheath /Delivery System Assembly.
Note: To inject contrast, a syringe or a manifold must be attached to the flush port of the Delivery System.
13. Before deploying the occluder, confirm the position of the delivery catheter tip using fluoroscopy and transesophageal echocardiography (TEE).
14. If repositioning is required, loosen the delivery catheter and carefully withdraw it from the guiding sheath. If needed, reinsert the pig-tail catheter to reposition the guiding sheath. Then, reinsert the delivery catheter as described in steps 11 and 12.
15. Release the Occluder by loosening the valve on the Delivery System, ensuring that the handle remains stable during withdrawal of the Delivery System, and maintaining the connection of the Core Wire.
16. Occluder release criteria: Position, Anchor, Size, and Seal (PASS criteria):
A. Position: Plane of maximum diameter of the Occluder should be at or just distal to the LAA stium, where possible (see Figure 1), while meeting all other PASS criteria.
B. Anchor: Move the tip of the Guiding Sheath back to expose sufficient core wire. Next, gently pull back on the deployment knob to visualize movement of the Occluder and LAA together.
C. Size (compression): Measure plane of maximum diameter of Occluder . Use Table 1 as a guide.
D. Seal: Ensure all lobes are distal to Occluder and sealed (i.e., no leak > 5 mm).

Figure 1 .LeSifterTM Device Position and Size
Note: If repositioning of the Occluder is required, proceed to Step 17 (Occluder repositioning). If removal of the Occluder is required, proceed to Step 18 (Occluder recapture and removal). If the Occluder meets release criteria, proceed to Step 19 ( Occluder release).
17. If repositioning of the Occluder is required, recapture the Occluder per the following instructions.
A. Advance tip of Guiding Sheath /Delivery System Assembly up to Occluder and align Guiding Sheath/Delivery System Assembly with Occluder (do not unsnap).
B. Fix deployment knob position with right hand and gently advance Guiding Sheath / Delivery System Assembly over the Occluder by positioning the right thumb against the Delivery System hemostasis valve for stability and push the right thumb forward. Resistance will be felt as the Occluder is collapsed into the Delivery System.
C. Continue to advance the Guiding Sheath /Delivery System Assembly such that the Occluder anchors are released from the LAA. The Occluder may be fully recaptured into the Guiding Sheath /Delivery System Assembly as needed prior to redeployment. After recapture, the Occluder may be repositioned using the guidance found in Step 13.
18. If removal of the Occluder is required, recapture the Occluder per the following instructions.
A. Advance tip of Guiding Sheath /Delivery System Assembly up to face of Occluder (do not unsnap).
B. Fix deployment knob position with right hand and gently advance Guiding Sheath / Delivery System Assembly over the Occluder by positioning the right thumb against Delivery System hemostasis valve for stability and push the right thumb forward. Resistance will be felt as the Occluder is collapsed into the Delivery System. Continue to advance the Assembly until Occluder is completely collapsed, recaptured, and distal fluoro tip is proximal to the Delivery System Distal Marker Band. Tighten the Delivery System hemostasis valve.
C. Ensure Guiding Sheath hemostasis valve is fully open.Unsnap Delivery System from Guiding Sheath while maintaining Guiding Sheath position.Slowly remove Delivery System.
D. Insert pigtail catheter to reposition Guiding Sheath in LAA,if necessary.
E. Repeat Steps 8-15 with new Delivery System.
19. If the Occluder meets release criteria,release Occluder .
Warning:Do not release the Occluder from the core wire if the Occluder does not meet all release criteria.
A.Confirm proper Occluder release criteria:Positon,Anchor,Size,and Seal(PASS criteria).
B.Advance Guiding Sheath/Delivery System Assembly close to face of Occluder .
C.Rotate deployment knob counterclockwise 3-5 full turns.
D.Confirm core wire is disconnected.
20. Ensure Guiding Sheath is free from anatomical structures prior to removal;adjust Guiding Sheath as necessary.Stop if resistance is felt.
21. Remove Guiding Sheath and Delivery System based on parameters for hemostasis.
22. Use standard of care for post-procedure bleeding at access site.
POST-PROCEDURE INFORMATION
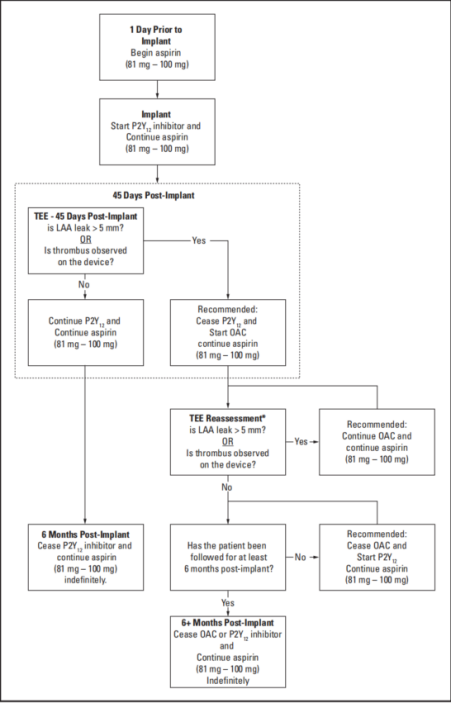
Options for post-procedure antithrombotic therapy are shown below. Physicians should exercise clinical judgement based on individual patient characteristics in determining the most appropriate use of antithrombotic drugs for the post-implant medication regimen.
Option A) Short-term OAC (see Figure 2)
1. Patients should remain on 81 mg - 100 mg of aspirin. OAC therapy should be added post-implant. At 45 days (± 15 days) post-implant, perform WATCHMAN FLX Pro Device assessment with TEE. Cessation of OAC therapy is at physician discretion provided that any leak demonstrated is ≤ 5 mm. If adequate seal is not demonstrated, subsequent OAC therapy cessation decisions are contingent on demonstrating leak is ≤ 5 mm. At the time the patient ceases OAC therapy, the patient should continue aspirin and begin a P2Y12 inhibitor daily. This regimen should continue until 6 months have elapsed after implantation. Patients should then remain on aspirin indefinitely. If a patient remains on OAC therapy and aspirin 81 mg - 100 mg for at least 6 months after implantation and then ceases OAC therapy, the patient should not require a P2Y12 inhibitor but should continue aspirin daily.
2. At 45 days and at 12 months, perform imaging to assess the WATCHMAN FLX Pro Device with TEE.
• Confirm absence of intra-cardiac thrombus.
• Perform color Doppler assessment to include the device/LAA border at the following approximate TEE angles (0°, 45°, 90° and 135°). Measure any residual leak around the device into the LAA. If there is evidence of leak > 5 mm, continuing or restarting antigoagulation therapy is recommended.
• If thrombus is observed on the device, use of anticoagulation is recommended until resolution of
thrombus is demonstrated by TEE.
3. Prescribe appropriate endocarditis prophylaxis for 6 months following Closure Device implantation.
The decision to continue endocarditis prophylaxis beyond 6 months is at physician discretion.
Option B) DAPT-only (see Figure 3)
1. Patients should remain on 81 mg – 100 mg of aspirin. P2Y12 inhibitor therapy should be added post-implant. At 45 days (± 15 days) post-implant, perform WATCHMAN FLX Pro Device assessment with TEE. P2Y12 inhibitor therapy should continue for 6 months provided that any leak demonstrated is ≤ 5 mm. If adequate seal is not demonstrated, discontinuation of P2Y12 and starting OAC is recommended. Subsequent OAC therapy cessation decisions are contingent on demonstrating leak is ≤ 5 mm. At the time the patient ceases OAC therapy, the patient should continue aspirin and re-start P2Y12 inhibitor daily. This regimen should continue until 6 months have elapsed after implantation. Patients should then remain on aspirin indefinitely.
2. At 45 days and at 12 months, perform imaging to assess the WATCHMAN FLX Pro Device with TEE.
• Confirm absence of intra-cardiac thrombus.
• Perform color Doppler assessment to include the device/ LAA border at the following approximate TEE angles (0°, 45°, 90° and 135°). Measure any residual leak around the device into the LAA. If there is evidence of leak > 5 mm, discontinuation of P2Y12 and starting OAC is recommended.
• If thrombus is observed on the device, use of anticoagulation is recommended until resolution of thrombus is demonstrated by TEE.
3. Prescribe appropriate endocarditis prophylaxis for 6 months following Closure Device implantation.
The decision to continue endocarditis prophylaxis beyond 6 months is at physician discretion.

Link to sscp (summary of safety and clinical performance)with product basic UDI-DI
Clinical Benefit
Clinical Trial Summary
Objective of the Trial
The trial aimed to assess the safety and effectiveness of the LAA occluder system in preventing thromboembolic events originating from the LAA in patients with non-valvular atrial fibrillation (NVAF) who are unsuitable for long-term anticoagulant therapy but suitable for short-term anticoagulation.
Before market release, this product was evaluated at six centers. Planned enrollment was 183 subjects, with 184 actually recruited, all of whom underwent left atrial appendage (LAA) occlusion therapy. The results provide supporting clinical data for product registration in the domestic market.
Trial Design
The trial followed a prospective, multicenter, single-arm target-value design, evaluating the safety and effectiveness of LAA occluder placement via the femoral vein in subjects at high risk of stroke (CHA2DS2-VASc ≥ 2) who are not suitable candidates for long-term anticoagulant therapy such as warfarin.
Target Values
The target thresholds for primary endpoints were as follows:
|
Primary Endpoint |
Target Value |
Expected Achievement Rate |
|
12-month ischemic stroke rate |
<1.3% |
<6% |
|
12-month LAA closure rate |
>93.6% |
>89.5% |
Sample Size
Based on calculations with a one-sided α of 0.025, 143 subjects provide at least 80% power to observe a 12-month ischemic stroke rate below 6%, while 142 subjects provide at least 80% power to observe a 12-month LAA closure rate above 93.6%. With an estimated 20% dropout rate, the sample size was set at the larger calculated value, resulting in a planned recruitment of 179 subjects for the clinical trial. The sample size calculation formula is as follows:
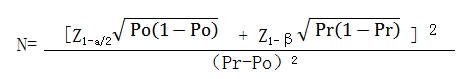
Po: Target value
Pr: Expected performance metrics of the device (ischemic stroke rate, LAA closure rate)
Inclusion Criteria
All enrolled subjects had to meet the following criteria:
• Age between 18 and 80 years with NVAF (excluding rheumatic mitral disease), CHA2DS2-VASc score ≥ 2.
• Indicated for long-term anticoagulation but unsuitable for such therapy due to one of the following conditions:
• Documented history or tendency of bleeding (e.g., gastrointestinal or cerebral bleeding).
• High risk of bleeding (HAS-BLED score ≥ 3).
• Poor compliance or intolerance to anticoagulants like warfarin.
• Compliance with short-term postoperative oral warfarin and aspirin + clopidogrel.
• The subject or legal representative understands the study’s nature, agrees to all terms, signs an ethics-approved informed consent form, and consents to the postoperative treatment plan and follow-up requirements.
Exclusion Criteria
1.Clinical Exclusion Criteria:
• Clinical Diagnoses:
• Conditions requiring long-term anticoagulation other than AF (e.g., recurrent pulmonary embolism or other deep vein thrombosis).
• Mitral stenosis (valve area <1.5 cm²), history of mitral balloon valvuloplasty, or mechanical valve replacement.
• Other valvular diseases requiring surgical intervention.
• Temporary AF due to surgical or interventional procedures.
• Presence of mural thrombus in the left atrium or LAA.
• Symptomatic carotid disease (e.g., carotid stenosis >50%).
• Recent myocardial infarction (<3 months) or diagnosis of non-ST elevation acute coronary syndrome or significant myocardial ischemia with revascularization <30 days.
• NYHA class IV heart failure.
• New ischemic stroke or TIA within the past 30 days.
• Active infection or sepsis, serious infectious disease within one month prior to surgery, malignancy.
• Bleeding disorder or active ulcers (e.g., cerebral hemorrhage <6 months, gastrointestinal ulcer bleeding <3 months).
• Active endocarditis, intracardiac vegetation, or other infections causing bacteremia.
• Cardiac or other tumors with an expected life expectancy of less than two years.
• Laboratory Findings:
• Hematological abnormalities (e.g., WBC <3×10⁹/L, acute anemia HB <90g/L, platelet count <100×10⁹/L), bleeding tendency or history of coagulopathy.
• Renal insufficiency (creatinine >3.0 mg/dL, or requiring dialysis).
• Device-Related:
• Prior LAA occlusion or surgical LAA removal.
• Prior heart transplant, septal repair, or septal occluder placement.
• Patients should avoid radiofrequency ablation within 30 days after LAA occlusion (risk of occluder displacement or thrombosis).
• Patients should avoid cardioversion within 30 days after LAA occlusion (risk of occluder displacement or thrombosis).
• History of mechanical heart valve replacement.
• Allergic or contraindicated to nitinol, aspirin, clopidogrel, or heparin.
• Planned cardiac or non-cardiac surgery within 30 days post-occlusion.
Others:
• Pregnancy or planning to conceive during the trial.
• Participation in other drug or medical device trials within 30 days prior to screening.
• Poor compliance or inability to complete the study according to the investigator’s judgment.
2. Transthoracic and Transesophageal Ultrasound Exclusion Criteria:
• LAA orifice diameter <14 mm or >32 mm on transesophageal echocardiography.
• LVEF <30%.
• Presence of confirmed or suspected thrombus on pre-implantation transesophageal echocardiography.
• Significant mitral stenosis (valve area <1.5 cm²) or other non-mitral lesions requiring surgery or cardiac tumors.
• Significant and unexplained pericardial effusion (≥4 mm).
Evaluation Metrics
1.Primary Endpoints
• Ischemic stroke rate within 12 months post-surgery (events/patient-year).
• LAA closure rate at immediate post-operation, 2 months, 6 months, and 12 months post-surgery (events per number of cases receiving occluder implantation).
2.Secondary Endpoint
• The perioperative complication rate observed within 7 days post-operation or before discharge. Complications include death, stroke, non-fatal myocardial infarction, cardiac tamponade/significant pericardial effusion, major bleeding requiring clinical intervention (intracranial or gastrointestinal bleeding, or any bleeding requiring 2 units of packed red blood cell transfusion), and puncture site complications (such as arteriovenous fistula repair, unplanned vascular grafting, major vessel repair in the chest or abdomen, or infection at the puncture site).
• The composite event rate within 12 months post-operation, including mortality (cardiac-related, non-cardiac-related, or unknown cause), stroke (ischemic, hemorrhagic, or unspecified), ischemic stroke/TIA/systemic embolism, major bleeding (intracranial or gastrointestinal, or any bleeding requiring 2 units of packed red blood cell transfusion), device-related complications requiring intervention (such as cardiac tamponade/significant pericardial effusion, device dislodgement), among others.
Trial Results
1.Nrollment and Baseline Data
This trial was conducted across six centers, screening a total of 199 participants. Of these, 12 did not proceed due to screening failure (5 participants failed screening criteria, and 7 withdrew informed consent). A total of 184 participants underwent left atrial appendage occlusion, with 176 completing the trial. Eleven participants discontinued early: 3 due to adverse events, 3 were lost to follow-up, and 5 withdrew for other reasons. Three female participants had a CHA2DS2-VASc score of 2. All 184 participants who underwent the procedure and used the trial device were included in the Full Analysis Set (FAS), Per-Protocol Set (PPS), and Safety Set (SS).
2.Efficacy Results Primary Endpoint
Table 2 statistical analysis results for the primary endpoint.
|
Evaluation Indicators |
FAS |
PPS |
|
(N=184) |
(N=184) |
|
|
Incidence rate of ischemic stroke within 12 months post-procedure (events per patient-year) |
0.6 % (1/179) |
0.6 % (1/179) |
|
Immediate left atrial appendage closure rate post-procedure |
98.4 %(181/184) |
98.4 %(181/184) |
|
Left atrial appendage closure rate at 2 months post-procedure |
100 %(158/158) |
100 %(158/158) |
|
Left atrial appendage closure rate at 6 months post-procedure |
100 %(151/151) |
100 %(151/151) |
|
Left atrial appendage closure rate at 12 months post-procedure |
100%(181/181) |
100%(181/181) |
Secondary Endpoint
Table 3 statistical analysis results for the secondary endpoint.
|
Evaluation Indicators |
FAS (N=184) |
PPS (N=184) |
|
Incidence of perioperative complications before discharge or within 7 days post-procedure (%) |
2.2% |
2.2 % |
|
Puncture site complications (%) |
0.5%(1/184) |
0.5%(1/184) |
|
Major bleeding requiring clinical intervention (%) |
1.6%(3/184) |
1.6%(3/184) |
|
Cardiovascular-related complications (%) |
1.1%(2/184) |
1.1%(2/184) |
|
Mortality (%) |
1.1%(2/184) |
1.1%(2/184) |
|
Cardiogenic death |
0.5%(1/184) |
0.5%(1/184) |
|
Non-cardiogenic death |
0.5%(1/184) |
0.5%(1/184) |
|
Death of unknown cause |
0 |
0 |
|
Stroke, n (%) |
0 |
0 |
|
Device dislocation, n (%) |
0.5%(1/184) |
0.5%(1/184) |
|
Incidence of Composite Events at 12 Months Post-Procedure (%) |
6.0 % |
6.0 % |
|
Major bleeding, n (%) |
2.7%(5/184) |
2.7%(5/184) |
|
Intracranial hemorrhage |
0 |
0 |
|
Gastrointestinal bleeding |
2.2%(4/184) |
2.2%(4/184) |
|
Any bleeding requiring 2 units of red blood cell transfusion |
0.5%(1/184) |
0.5%(1/184) |
|
Cardiovascular-related complications, n (%) |
1.6%(3/184) |
1.6%(3/184) |
|
Non-fatal myocardial infarction |
0 |
0 |
|
Arrhythmia other than atrial fibrillation |
0 |
0 |
|
Cardiac tamponade/significant pericardial effusion |
1.1%(2/184) |
1.1%(2/184) |
|
Cardiogenic shock |
0 |
0 |
|
Endocarditis |
0 |
0 |
|
Other |
0.5%(1/184) |
0.5%(1/184) |
|
Systemic embolism, n (%) |
0.5%(1/184) |
0.5%(1/184) |
|
Cardiac |
0 |
0 |
|
Other (specify) |
0.5%(1/184) |
0.5%(1/184) |
|
Mortality, n (%) |
2.7%(5/182) |
2.7%(5/182) |
|
Cardiogenic death |
0.5%(1/184) |
0.5%(1/184) |
|
Non-cardiogenic death |
0.5%(1/184) |
0.5%(1/184) |
|
Death of unknown cause |
0 |
0 |
|
Stroke, n (%) |
0.5%(1/184) |
0.5%(1/184) |
|
Ischemic |
0.5%(1/184) |
0.5%(1/184) |
|
Hemorrhagic |
0 |
0 |
|
Unspecified |
0 |
0 |
|
Transient ischemic attack (TIA) |
0 |
0 |
|
Device dislocation, n (%) |
0.5%(1/184) |
0.5%(1/184) |
|
|
|
|
|
Occluder Performance |
|
|
|
mmediate Post-Procedure (Surgical Record) |
|
|
|
No dislocation in device tug test |
100 %(182/182) |
100 %(182/182) |
|
Complete left atrial appendage (LAA) occlusion, n (%) |
98.3%(176/179) |
98.3%(176/179) |
|
Device-related thrombus |
0 %(0/182) |
0 %(0/182) |
|
Post-Procedure to Discharge Visit |
|
|
|
Device in position |
100 %(175/175) |
100 %(175/175) |
|
New device-related thrombus |
0 %(0/175) |
0 %(0/175) |
|
30 Days Post-Procedure ± 7 Days |
|
|
|
Device in position |
100 %(125/125) |
100 %(125/125) |
|
New device-related thrombus |
0 %(0/125) |
0 %(0/125) |
|
2 Months Post-Procedure ± 15 Days |
|
|
|
Device in position |
100 %(158/158) |
100 %(158/158) |
|
Residual LAA shunt (via transesophageal Doppler or chest CT) |
16.5 %(26/1158) |
16.5 %(26/158) |
|
Residual shunt grade 2-5 |
100%(26/26) |
100%(26/26) |
|
New device-related thrombus |
1.9 %(3/158) |
1.9 %(3/158) |
|
6 Months Post-Procedure ± 15 Days |
|
|
|
Device in position |
100 %(151/151) |
100 %(151/151) |
|
Residual LAA shunt (via transesophageal Doppler or chest CT) |
14.6 %(22/151) |
14.6 %(22/151) |
|
Residual shunt grade 2-5 |
100%(22/22) |
100%(22/22) |
|
New device-related thrombus |
0.7 %(1/151) |
0.7 %(1/151) |
|
12 Months Post-Procedure ± 30 Days |
|
|
|
Device in position |
100 %(154/154) |
100 %(154/154) |
|
New device-related thrombus |
0 %(0/154) |
0 %(0/154) |
|
Device Procedural Success Rate (%) |
98.4 % |
98.4 % |
|
Loading of occluder into delivery system, n (%) |
100 %(184/184) |
100 %(184/184) |
|
Successful delivery to LAA via delivery system, n (%) |
100 %(184/184) |
100 %(184/184) |
|
Accurate device release, n (%) |
98.4 %(181/184) |
98.4 %(181/184) |
|
Complete retraction of delivery system, n (%) |
100 %(184/184) |
100 %(184/184) |
|
|
|
|
|
Complete Device Release Rate (%)
95% Confidence Interval (exact probability method) |
72.6 % |
72.6% |
|
No significant shoulder protrusion |
76.2 %(138/181) |
76.2%(138/181) |
|
No dislocation in tug test post-release |
100%(184/184) |
100%(184/184) |
|
Compression ratio post-release between 10-30% |
97.8 %(175/179) |
97.8 %(175/179) |
|
Complete LAA occlusion |
98.3%(176/179) |
98.3%(176/179) |
|
|
|
|
|
Technical Success Rate (%) |
98.4 % |
98.4 % |
3.Table 3 Summary of Adverse Events
|
Adverse Event |
N=184 |
|
|
|
Number of Occurrences |
Number of Cases(%) |
|
Adverse Event |
188 |
92 ( 50.0) |
|
Serious Adverse Event |
53 |
36 ( 19.6) |
|
Adverse Event Related to the Investigational Device |
5 |
5 ( 2.7) |
|
Adverse Event Related to the Surgery |
18 |
13 ( 7.1) |
|
Serious Adverse Event Related to the Investigational Device System |
2 |
2 ( 1.1) |
|
Adverse Event Leading to Withdrawal |
3 |
3 ( 1.6) |
|
Adverse Event Leading to Death |
7 |
5 ( 2.7) |
Transportation and Storage Conditions
The product should be protected from moisture, direct sunlight, rain, high temperatures, heavy pressure, and impact during transportation. The product should be stored in a cool, dry place, away from direct light.
Shelf Life
Three years
Warranty
Nanjing YDB Medical Technology Co., Ltd. (YDB) guarantees that the design and manufacture of this product have been thoroughly considered. This warranty supersedes and excludes all other warranties not explicitly stated herein, whether express or implied by law, including but not limited to any implied warranties of merchantability or fitness for a particular purpose. The handling, storage, cleaning, disinfection, and other factors related to the patient, diagnosis, treatment, surgery, and any other issues beyond YDB’s control can directly affect the product and its performance. Under this warranty, YDB’s obligations are limited to the repair or replacement of the product, and YDB is not liable for any incidental or consequential losses, damages, or costs arising from the direct or indirect use of this product. YDB will not assume or authorize any other person to assume any additional or other responsibilities or obligations in relation to this device. YDB does not bear any responsibility for the reuse, reprocessing, or re-sterilization of this device, nor does it make any express or implied warranties related to the device, including but not limited to warranties of merchantability or fitness for a particular purpose.
Appendix A: Supplemental Information
|
REF of Device |
Occluder |
Access System |
||||
|
D(mm) |
L(mm) |
Delivery System |
Guiding System |
|||
|
Core Wire(mm) |
Delivery Sheath(mm) |
Guiding Sheath(mm) |
DIlation Tube(mm) |
|||
|
YFDQ-20-1 |
20 |
11 |
945 |
800 |
760 |
817 |
|
YFDQ-20-2 |
/ |
/ |
||||
|
YFDQ-24-1 |
24 |
13 |
760 |
817 |
||
|
YFDQ-24-2 |
/ |
/ |
||||
|
YFDQ-27-1 |
27 |
15 |
760 |
817 |
||
|
YFDQ-27-2 |
/ |
/ |
||||
|
YFDQ-31-1 |
31 |
17 |
760 |
817 |
||
|
YFDQ-31-2 |
/ |
/ |
||||
|
YFDQ-35-1 |
35 |
19.5 |
760 |
817 |
||
|
YFDQ-35-2 |
/ |
/ |
||||
|
CE |
REP |
SUNGO Europe B.V.
Fascinatio Boulevard 522,Unit 1.7,2909VA
Capelle aan den IJssel ,The Netherlands
Nanjing YDB Medical Techonology Co.,Ltd.
No.129, Fengshan Road, Gaochun Economic Development Zone, Nanjing,Jiangsu,China
Tel: +86 25 56865078
Fax: +86 25 56865079
Web:www.ydbmed.com